
Records and data looking by investigator at the time of audit?
By QEdge Team
Published On February 29, 2020
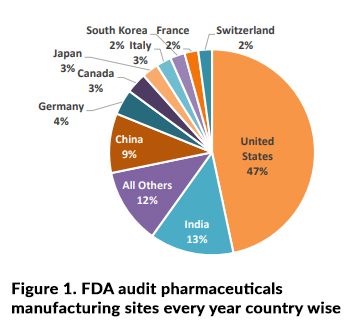
FDA audits conduct at various pharmaceutical manufacturing sites according to Figure 1 data within entire globe and investigators looking for typically, proof of compliance with current Good Manufacturing Practices (GMP) with respective to patient safety. In the case of pharmaceuticals, this will relate to part & sub part of the 21 Code of Federal Regulations (CFR) Parts 210 and 211 as below.
- Facility & Equipments: 21 CFR part 211, subpart B, C, D & J
- Material system: 21 CFR part 211, subpart B, E, H & J
- Packaging & labelling system: 21 CFR part 211, subpart B, G & J
- Laboratory control system: 21 CFR part 211, subpart B, I, J & K
This can involve looking at whether thorough responses are made in the incidence of typical events, which might include Product review, complaint review, discrepancies and failure investigation, related to manufacturing, packaging, testing, change control, product improvement project, reprocess/ rework, returns/salvages, stability failure, quarantine products, validations, QC unit functions, training, qualification of employees, change requests, deviations, specific failures, rejections, complaints and effectiveness of CAPA. Here, investigators will be checking that issues are dealt with appropriately.
Investigators will look at the appropriateness of quality processes, whether they are robust and consistent, and whether complaints or failures in the market could indicate that a process might be questionable. Real time monitoring and the use of quality systems to provide an evidence of what is happening within a process, will also be evaluated. To meet requirements, manufacturers must demonstrate that they have a system in place that will monitor the quality of a product in real time, and which empowers them to respond to issues quickly. Finally, the investigator will be looking to establish trust in a manufacturer’s data integrity, particularly the validity and transparency of shop floor data and laboratory data otherwise insufficient data would be persist in Audit observation.
Types of FDA Inspection & Audit
Each FDA Inspection/Audit is intended to help protect the public from unsafe products, but the focus and expectations of each type of inspection are different.
(a) Pre-Approval Inspections: conducted after a company submits an application to FDA to market a new product.
(b) Routine Inspections: mandated by law every 2 years once (for class II and class III medical device manufacturers, nutraceutical, cosmetic, food and drug manufactures)
(c) Compliance Follow-Up Inspections: to review actions taken by a manufacturer in response to a previous inspection that resulted in significant FDA 483 audit observations or a Warning Letter.
(d) For Cause Inspections: to investigate a specific problem that has been reported to FDA. The source of the report can be from the manufacturer or consumer or user or even an employee in the same company.
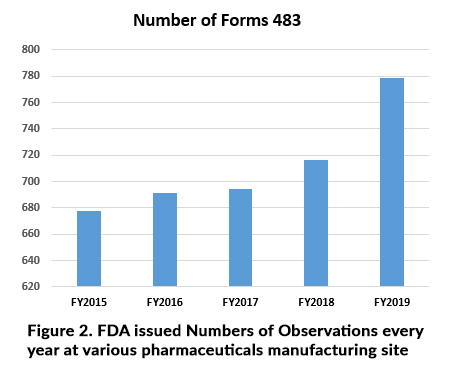
Based on above all various types of inspections may lead to observation at manufacturing site as per Figure 2. reflect every year it should be goes rise and Problems may indicate: Filed alert report, (FARs) Industries complaint, recalls, Indicators of defective products and warning letters.
The role of QEdge technology
QEdge deliver quite another way to prevent audit observations that start FDA 483 is through the implementation of QEdge technology which can automate many aspects of quality processes. This allows manufacturing organizations to ensure controlled quality processes, flow management and effectiveness checking, as well as enabling users’ activities for monitoring and reporting purposes. therefore, this type of system can help manufacturers achieve compliance improvements, performance improvements and real time visibility of data.
Conclusion
With regulations increasing in complexity, the use of robust QEdge solution in combination with real time monitoring of processes and problems, is vital in preventing audit observation FDA 483. The technology is now being adopted at a faster rate in the life sciences space than any other drug manufacturing domain. Despite this, the fact remains that all too often operations managers find the prospect of implementing new processes daunting. The benefits might be clear, but there is a perception that QEdge systems are implemented on plant server as well as cloud-based systems have made cost effective solutions accessible, knocking the prospect to improve quality and increase compliance within the reach of organizations of all sizes.
